
The Profile for Cystic Fibrosis
The Profile for Cystic Fibrosis
Cystic fibrosis is an inherited disease that occurs when both parents carry the mutant CF gene. It is incurable but new advances are being found each year in finding new methods of treatments. New methods for diagnosing the disease are being discovered as well. The disease causes the normally thin mucus that lines the passageways to major organs to become thick and sticky causing it to build up and cause problems for the patient in breathing and digesting their food. The disease can also affect the liver, bones, and the reproductive organs. The lifespan of a cystic fibrosis patient has risen from 16 years old to somewhere in the middle 30’s. Symptoms will vary for each person diagnosed with the disease.
The Profile for Cystic Fibrosis
CF is caused by mutations in the cystic fibrosis transmembrane conductance regulator gene. A person can only inherit this disease if both parents are carriers of this mutant gene. Someone may be a carrier of the mutant gene but show no signs of the disease. This carrier may still be able to pass the disease to their children if their partner also carries the mutant gene.
The Cystic Fibrosis Foundation gathered the statistics of people who have cystic fibrosis. They estimate 30,000 Americans, about 20,000 Europeans, and 3,000 Canadians have cystic fibrosis. Strangely enough, if you are a Caucasian who had family originating in Northern Europe, you are more at risk for carrying, having, and passing on the disease. Ethnic groups such as African-Americans, Native Americans, and Asians have a reduced risk of developing cystic fibrosis. The Cystic Fibrosis Foundation estimates there are over twelve million people may have the mutant gene but are unaware they are carriers.
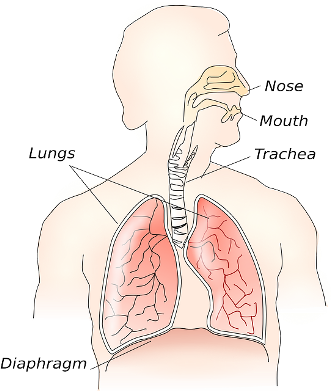
The symptoms of cystic fibrosis do not always follow the same pattern for each patient. It affects each person in a different way and in different degrees. Cystic fibrosis had one basic issue, an abnormality in glands that produce sweat and mucus. Sweat is important to keep the body cool and mucus lines the reproductive, digestive, and respiratory organs preventing them from drying out. The normally thin mucus helps protect these important organs from infections. When the mucus becomes too thick and sticky it lodges in the passageways and are receptors for any infection or bacteria that enters the system.
Cystic fibrosis patients lose an excessive amount of salt in sweat and this upsets the balance of minerals in the blood. This can cause abnormal heart rhythms and shock is another possibility for CF patients.
Thick mucus can result in malnutrition, slow growth rate, frequent respiratory infections, and breathing difficulties. It can lead to permanent lung damage if not treated properly. Lung disease is the main cause of death in cystic fibrosis patients. Other parts of the body may be affected by cystic fibrosis including the sinus cavities where polyps may form and the patient may have frequent sinus infections. The digestive tract may show signs of the disease with discomfort in the stomach, bloating, and excess gas.
